Дечак са ретком болешћу задивио лекаре после прве генске терапије на свету

Аутор фотографије, Chu family
- Аутор, Фергус Волш
- Функција, ББЦ, медицина
- Време читања: 10 мин
Трогодишњи дечак задивио је докторе напретком пошто је постао први на свету који је примио револуционарну нову генску терапију.
Оливер Чу има ретко, наследно стање звано Хантеров синдром или МПСИИ које изазива прогресивно оштећење тела и мозга.
Код најтежих случајева, пацијенти са овом болешћу обично умиру пре 20. године.
Хантеров синдром се понекад описује као нека врста дечје деменције.
Због фаличног гена, пре третмана Оливер није могао да производи ензиме кључне за одржавање ћелија здравим.
Први пут на свету, медицинско особље у Манчестеру покушало је да заустави ову болест мењајући Оливерове ћелије уз помоћ генске терапије.
„Двадесет година чекам да видим да је дечаку као што је Оли боље као што је сада њему и то је јако узбудљиво“, рекао је за ББЦ професор Сајмон Џонс, који је ко-предводник испитивања.
У средишту ове невероватне приче су Оливер, први од пет дечака на свету који је примио овај третман, и породица Чу, из Калифорније, који су положили сву веру у медицински тим Краљевске дечије болнице у Манчестеру.




Годину дана после отпочињања третмана, изгледа као да се Оливер сада развија нормално.
„Сваки пут кад говоримо о томе, на ивици сам суза зато што је све сувише фантастично“, каже његова мајка Џингру.
ББЦ је пратио Оливерову причу више од годину дана, па и то како су научници у Великој Британији први пут развили пионирску генску терапију и како њихова медицинска испитивања замало нису била прекинута због недостатка средстава.
Погледајте видео: Синдром ЦДКЛ5 - живот родитеља деце са ретким болестима

Извлачење матичне ћелије - децембар 2024.
Први пут смо срели Оливера и његовог тату Рикија у децембру 2024. године у клиничкој установи за истраживања, у Краљевској дечјој болници у Манчестеру.
Био је то велики дан.
Откако је добио дијагнозу Хантеровог синдрома у априлу, Оливеров живот, као и његовог старијег брата Скајлера, који такође има ово стање, сводио се углавном на посете болници.
Скајлер је показао известан позни развој у говору и координацији, али је то испрва било објашњено тиме што је рођен током пандемије корона вируса.
Рики ми каже да је дијагноза његовог сина била потпуни шок.
„Кад сазнате за Хантеров синдром, прва ствар коју вам доктор каже је: 'Не идите на интернет и не тражите га зато што ћете наћи само најтеже случајеве и бићете веома, веома обесхрабрени'.“
„Али, као и сви, ви то ипак погледате и кажете себи: 'Ох, мој боже, зар ће се ово догодити с оба моја сина?'“.
Деца су рођена наизглед здрава, али око друге године почну да показују прве симптоме болести.
Они варирају и могу да обухватају промене физичких особина, укоченост удова и низак стас.
Може да дође и до оштећења у читавом телу, међу њима и на срцу, јетри и зглобовима и, у најтежим случајевима, може да доведе до менталног оштећења и прогресивног неуролошког пропадања.
Хантеров синдром се скоро увек јавља код дечака.
Изузетно је редак - погађа једну од 100.000 мушких беба на свету.
Све до сада, једини лек доступан за Хантеров синдром био је елапрас, који кошта око 340.000 евра по пацијенту годишње и може да успори физичке последице болести.
Лек не може да прескочи крвно-мождану баријеру и не помаже код когнитивних симптома.
Међутим, данас је Оливер повезан са машином и одстрањују му неке ћелије, што је први кључни корак у покушају да се заустави његов генетски поремећај у овом јединственом третману.
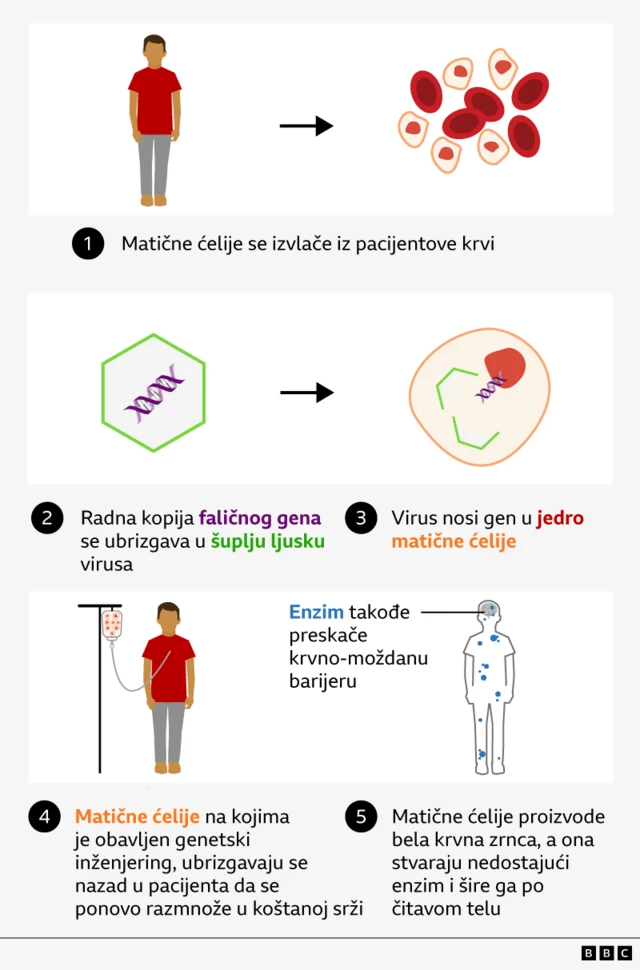
„Његова крв пролази кроз машину која сакупља конкретан тип ћелија - матичне ћелије - које ће бити послате у лабораторију на модификацију и потом ће му бити враћене“, објашњава докторка Клер Хорган, специјалисткиња за педијатријску хематологију.
Промена Оливерових ћелија
Оливерове ћелије се пажљиво пакују и шаљу у лабораторију у болницу Грејт Ормонд Стрит (ГОСХ) у Лондону.
Код Хантеровог синдрома, генетска грешка значи да ћелије немају инструкцију за прављење ензима, идуроната-2-сулфатазе (ИДС), кључног за разлагање великих молекула шећера који се временом накупе у ткиву и органима.
Научници убацују недостајући ген ИДС у вирус, чији је генетски материјал уклоњен тако да не може да изазове болест.
Сличан метод се користи и код других генских терапија, као што је третман другог наслеђеног стања - МЛД.
„Користимо машинерију из вируса да убацимо радну копију фаличног гена у сваку матичну ћелију", објашњава докторка Карен Бакленд, из Службе за ћелијску и генску терапију при светски познатој болници за делу Great Ormond Street Hospital (ГОСХ).
„Кад се то врати Оливеру, оне би требало да поново населе његову коштану срж и да почну да производе бела крвна зрнца, а свака од њих ће уз мало среће почети да производи недостајући протеин [ензим] у његовом телу.“
И даље остаје проблем како допремити довољно недостајућег ензима у мозак.
Да би то било решено, убачени ген је модификован тако да ензим ког производи ефикасније прескаче крвно-мождану баријеру.
Дан инфузије - фебруар 2025
Следећи пут се срећемо са Оливером поново у клиничкој установи за истраживање у Краљевској дечјој болници у Манчестеру.
Овај пут је с мајком Џингру, док је Рики остао у Калифорнији да се стара о Скајлеру.
Приметан је осећај ишчекивања док чланица истраживачког тима отвара велики метални резервоар за криопрезервацију у ком се налазе Оливерове замрзнуте матичне ћелије са измењеним генима, након што су допремљене из ГОСХ-а.
Мала, прозирна кеса за инфузију се извлачи напоље и полако доводи на телесну температуру на тацни пуној течности.
Након вишеструких провера, медицинска сестра увлачи прозирну течности која садржи 125 милиона матичних ћелија са модификованих геном у шприц.
Оливер је навикнут на болнице, али је немиран, и врпољи се док сестра полако убризгава третман количине једне шоље у катетер у његовим грудима.
Џингру држи Оливера чврсто у наручју.
После 10 минута, инфузија је обављена.
Сат времена касније иде друга, идентична инфузија.
Оливер наставља да гледа цртане филмове на преносивом екрану, несвестан потенцијалне важности онога што се управо догодило.
И то је то.
Генска терапија је завршена.
Чини се да је све готово прилично брзо, али је амбиција ипак огромна: зауставити Оливерову прогресивну болест у месту, у једнократном, јединственом третману.
После неколико дана, Оливер и Џингру лете назад за Калифорнију.
Сада породица и медицински тим морају да чекају да виде да ли је терапија успела.
Рани знаци напретка - мај 2025.
У мају, Оливер је поново у Манчестеру за кључна тестирања како би се видело да ли генска терапија функционише.
Овај пут је читава породица ту.
Срећемо се у парку у центру Манчестера и одмах је јасно да су изгледи врло добри.
Оливер је покретнији и љубопитљивији него што сам га икад видео.
У реду, он сада има слободу да се игра и више није у болници, али свакако делује ведрије и здравије.
Рики је узбуђен.
„Заиста му добро иде. Приметили смо напредак у говору и покретљивости. Сазрео је за само три месеца.“
Истински крупна вест је да Оливер не мора више да прима недељну инфузију недостајућег ензима.
„Желим да се уштинем сваки пут кад кажем људима да Оливер прави сопствене ензиме“, каже Џингру.
„Сваки пут кад разговарамо о томе на ивици сам суза зато што је то фантастично.“
Она ми каже да је „много другачији“ него пре третмана, говори „све време“ и више се упушта у интеракцију са другом децом.

Дивно је коначно срести петогодишњег Скајлера који је веома заштитнички настројен и брижан према млађем брату.
„Моја жеља је да и Скајлер може да добије исти третман“, каже Рики.
„Осећамо се као да је Оливер добио ресет у животу и ми сада желимо исто и за Скајлера, иако је он мало старији.“
Испрва се мислило да је Оливер сувише стар за испитивање, јер третман не може да исправи већ настала оштећења, али су тестови показали да је он још увек остао углавном нетакнут.
Скајлер делује као да ужива у свету око себе, радо ме држи за руку и ћаска са мном док шетамо по парку.
Рики објашњава да је Скајлер заостао у говору и моторичким способностима, али се подвргава терапији инфузије, која уноси третман у његово тело, али не и у мозак.
Погледајте видео: Ретке болести - живот са атаксијом

'Вечно захвални'
Оливер се враћа у Манчестер свака три месеца на неколико дана, ради контролних тестова.
Крајем августа, додатне провере потврђују да генска терапија функционише.
Оливер очигледно напредује и сада је већ прошло девет месеци од окончања третмана.
Професор Џонс, ког Оливер зове Деда Мраз због његове беле браде, сија од среће: „Пре трансплантације, Оли уопште није правио ензиме, а сада их прави стотинама пута више од нормалне количине.
„Али оно што је још важније, можемо да видимо да му је боље, учи, зна нове речи и има нове вештине, креће се унаоколо много лакше.“
Међутим, професор Џонс исказује одређени степен подозривости.
„Морамо да будемо обазриви и да се не занесемо у узбуђењу око свега, али ствари су онолико добре колико уопште могу да буду у овом тренутку“, каже.
У башти на крову болнице, Оливер се игра са татом.
„Као да је потпуно другачије дете. Трчи свуда, не престаје да прича“, каже Рики.
„Будућност за Олија делује веома блиставо и уз мало среће то значи да ће и друга деца добити третман.“
Петорица дечака су пријављена за пробно испитивање из САД, Европе и Аустралије.
Ниједан није из Велике Британије, јер су овдашњи пацијенти добили дијагнозу прекасно да би се квалификовали.
Сви дечаци ће бити праћени најмање две године.
Ако испитивање буде проглашено успешним, болница и универзитет се надају да ће ући у партнерство са још једном биотехнолошком фирмом како би третман добио званичну дозволу.
Професор Џонс каже да се исти приступ генској терапији примењује и на друге генетске поремећаје.
У току су слична пробна испитивања у Манчестеру за МПС типа 1, илити Харлеров синдром, и МПС типа 3, или Санфилипов синдром.
Рики и Џингру кажу да су „вечно захвални“ манчестерском тиму зато што је омогућио да се Оливер укључи у испитивање.
Они кажу да су запањени напретком оствареним у последњих неколико месеци.
Оливер сада производи недостајући ензим и његово тело и мозак су здрави.
„Не желим да баксузирам, али имам утисак да све иде веома, веома добро“, каже Рики.
„Његов живот се више не своди на игле и посете болници. Његов говор, покретљивост и когнитивни развој су постали драстично бољи.“
„Није само спора, постепена крива како постаје старији, већ је све скочило експоненцијално од трансплантације.“
Испитивање које се замало није десило

Истраживачи са Универзитета у Манчестеру предвођени професором Брајаном Бигером провели су више од 15 година радећи на стварању генске терапије за Хантеров синдром.
Универзитет је 2020. године саопштио да је ушао у партнерство са малом америчком биотехнолошком компанијом Авробио, како би спровели клиничко испитивање.
Међутим, три године касније, компанија је вратила лиценцу универзитету, после лоших резултата из друге студије генске терапије и недостатка средстава.
Прво испитивање на људима, које ће ускоро помоћи Оливеру, било је у опасности још пре него што је уопште почело.
„Морали смо да деламо веома брзо да бисмо покушали да спасемо читаву идеју и да пронађемо другог спонзора и други извор финансирања“, каже професор Џонс.
Тада је ускочила британска медицинска истраживачка добротворна организација ЛајфАрк, обезбедивши средства у висини од 2,8 милиона евра.
Извршни директор доктор Сем Барел каже: „Огромни изазов за више од 3,5 милиона људи у Великој Британији, који живе са ретким стањима, јесте обезбедити им приступ ефикасном лечењу - тренутно 95 одсто нема никакакав третман.“
Породица Чу осећа олакшање што испитивање није било обустављено и сада се нада да ће и Скајлер једног дана имати користи од исте генске терапије као и његов брат.
„Ходао бих до краја света, унатрашке, напред, наглавачке, бос, само да будем сигуран да моја деца имају бољу будућност“, закључује Рики.
Додатно извештавање: Нат Рајт и Бриџеш Пател
ББЦ на српском је од сада и на Јутјубу, пратите нас ОВДЕ.
Пратите нас на Фејсбуку, Твитеру, Instagramу и Вајберу. Ако имате предлог теме за нас, јавите се на bbcnasrpskom@bbc.co.uk